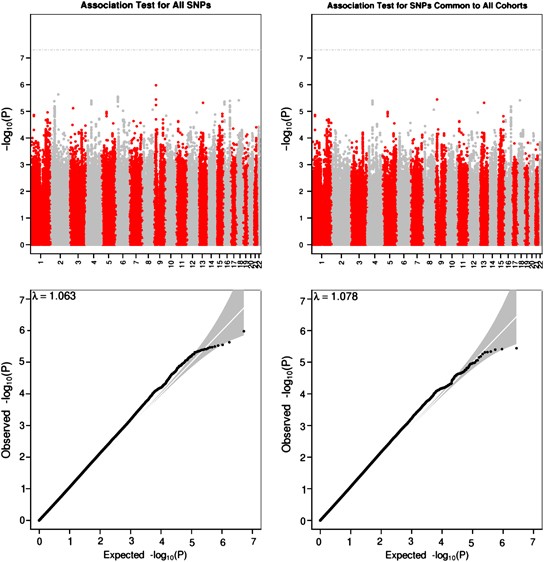
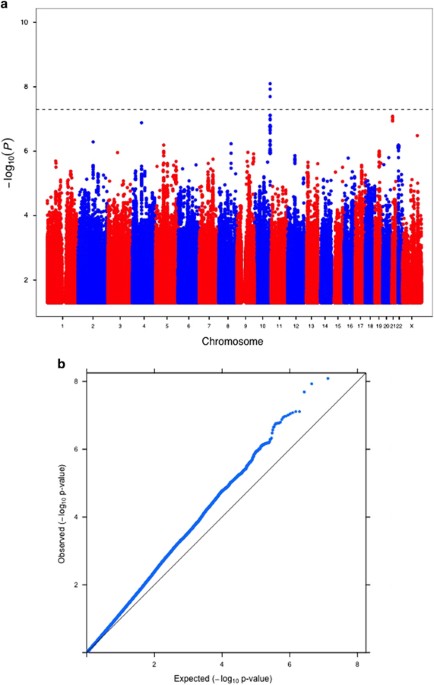
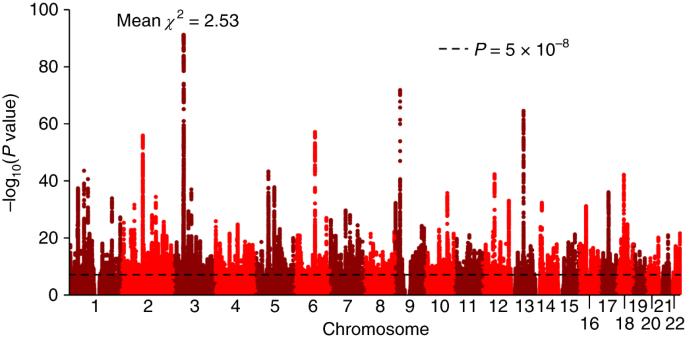
Intelligence in childhood, as measured by psychometric cognitive tests, is a strong predictor of many important life outcomes, including educational attainment, income, health and lifespan. Results from twin, family and adoption studies are consistent with general intelligence being highly heritable and genetically stable throughout the life course. No robustly associated genetic loci or variants for childhood intelligence have been reported. Here, we report the first genome-wide association study (GWAS) on childhood intelligence (age range 6–18 years) from 17 989 individuals in six discovery and three replication samples. Although no individual single-nucleotide polymorphisms (SNPs) were detected with genome-wide significance, we show that the aggregate effects of common SNPs explain 22–46% of phenotypic variation in childhood intelligence in the three largest cohorts (P=3.9 × 10−15, 0.014 and 0.028). FNBP1L, previously reported to be the most significantly associated gene for adult intelligence, was also significantly associated with childhood intelligence (P=0.003). Polygenic prediction analyses resulted in a significant correlation between predictor and outcome in all replication cohorts. The proportion of childhood intelligence explained by the predictor reached 1.2% (P=6 × 10−5), 3.5% (P=10−3) and 0.5% (P=6 × 10−5) in three independent validation cohorts. Given the sample sizes, these genetic prediction results are consistent with expectations if the genetic architecture of childhood intelligence is like that of body mass index or height. Our study provides molecular support for the heritability and polygenic nature of childhood intelligence. Larger sample sizes will be required to detect individual variants with genome-wide significance.