jeetcel
jeets me!
★★★★
- Joined
- Mar 21, 2023
- Posts
- 1,352
I was reading about some of these on Wikipedia, and oh man just read through common symptoms of said two genetic disorders or syndromes whatever
 en.wikipedia.org
en.wikipedia.org
The primary features are infertility and small, poorly functioning testicles (if present). These symptoms are often noticed only at puberty, although this is one of the most common chromosomal disorders, occurring in one to two per 1,000 live births.
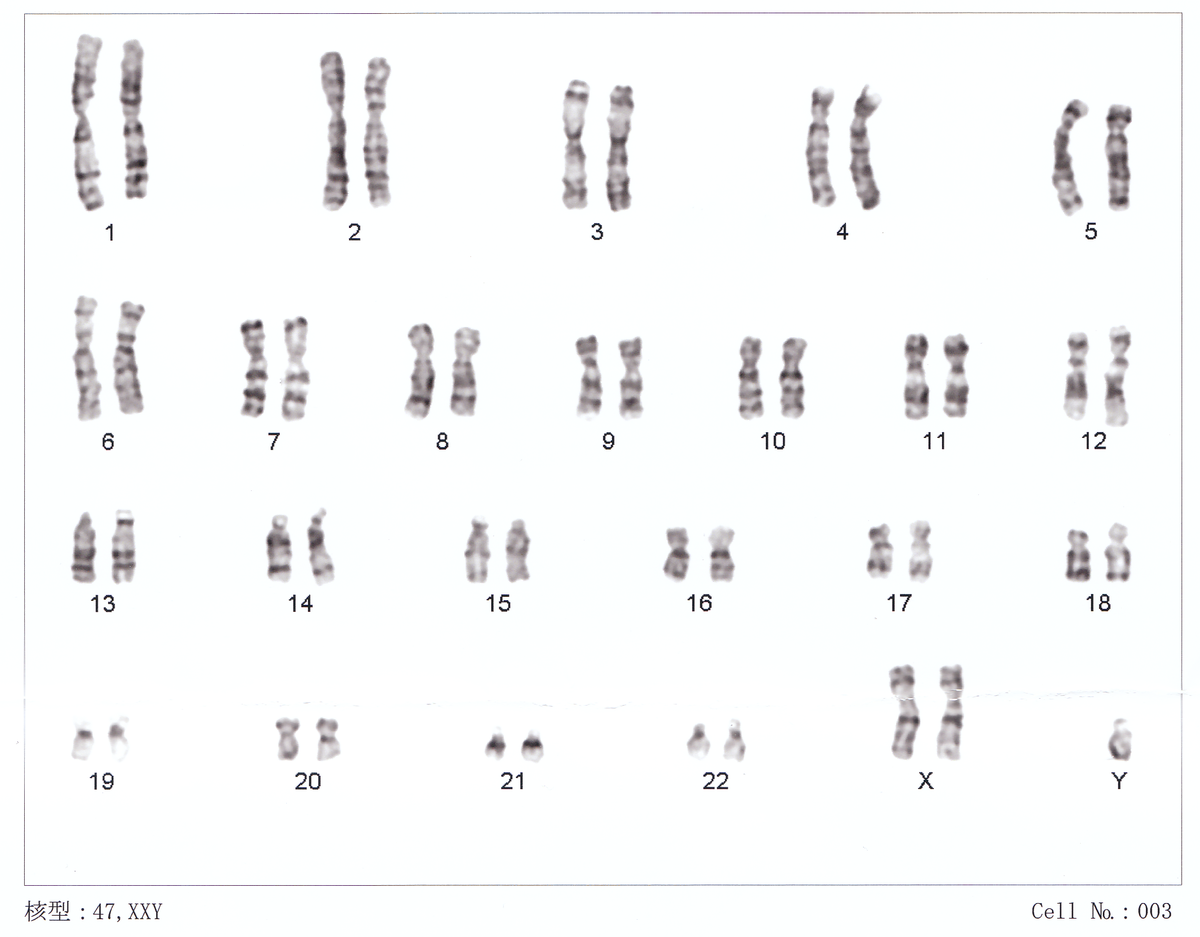
The syndrome is defined by the presence of at least one extra X chromosome in addition to a Y chromosome yielding a total of 47 or more chromosomes rather than the usual 46.
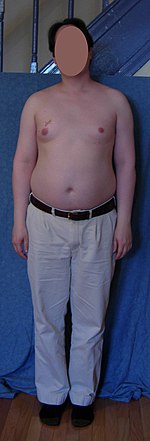
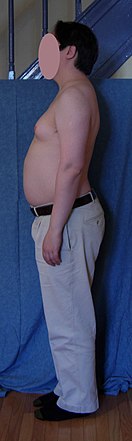
A person with typical untreated Klinefelter 46,XY/47,XXY mosaic, diagnosed at age 19 – a scar from biopsy is on his right breast above the nipple.
The Klinefelter syndrome has different manifestations and these will vary from one patient to another. Among the primary features are infertility and small, poorly functioning testicles. Often, symptoms may be subtle and many people do not realize they are affected. Whereas some other times symptoms are more prominent and may include weaker muscles, greater height, poor motor coordination, less body hair, gynecomastia (breast growth), and low libido. In the majority of the cases, these symptoms are noticed only at puberty.[10][6][4]
During puberty, they show less muscular body, less facial and body hair, and broader hips. This is a direct consequence of the low levels of testosterone produced by KS subjects. Delays in motor development may occur, which can be addressed through occupational and physical therapies. As teens, those assigned male at birth with XXY may develop breast tissue, have weaker bones, and a lower energy level than others. Testicles of those affected are usually less than 2cm in length (and always shorter than 3.5cm), 1cm in width, and 4ml in volume. Those with XXY chromosomes may also have microorchidism (i.e., small testicles).[15][16]
By adulthood, they tend to become taller than average; with proportionally longer arms and legs, less-muscular bodies, more belly fat, wider hips, narrower shoulders. Some will show little to no sign of affectedness, a lanky, youthful build and facial appearance, or a rounded body type with some degree of gynecomastia (increased breast tissue). Gynecomastia is present in approximately a third of affected individuals, a slightly higher percentage than in the XY population. Approximately 10% of those assigned male at birth with XXY chromosomes have gynecomastia noticeable enough that they may choose to have surgery. Those affected are often infertile, or have reduced fertility. Advanced reproductive assistance is sometimes possible in order to produce an offspring since approximately 50% of those assigned male at birth with Klinefelter syndrome can produce sperm.[17][18]
The overall IQ tends to be lower than average. Language milestones may also be delayed, particularly when compared to other people their age. Between 25% to 85% of those assigned male at birth with XXY have some kind of language problem, such as delay in learning to speak, trouble using language to express thoughts and needs, problems reading, and trouble processing what they hear. They may also have a harder time doing work that involves reading and writing, but most hold jobs and have successful careers.[15][21]
In 1995 a scientific study evaluated the psychosocial adaptation of 39 adolescents with sex chromosome abnormalities. It demonstrated that those assigned male at birth with XXY tend to be quiet, shy and undemanding; they are less self-confident, less active, and more helpful and obedient than other children their age. They may struggle in school and sports, meaning they may have more trouble fitting in with other kids.[21][26]
As adults, they live lives similar to others without the condition; they have friends, families, and normal social relationships. Nonetheless, some individuals may experience social and emotional problems due to problems in childhood. They show a lower sex drive and low self esteem, in most cases due to the feminine characteristics that their bodies display.[10][21]
In contrast to these potentially increased risks, rare X-linked recessive conditions are thought to occur less frequently in those with XXY than in those without, since these conditions are transmitted by genes on the X chromosome, and people with two X chromosomes are typically only carriers rather than affected by these X-linked recessive conditions.[30]
 en.wikipedia.org
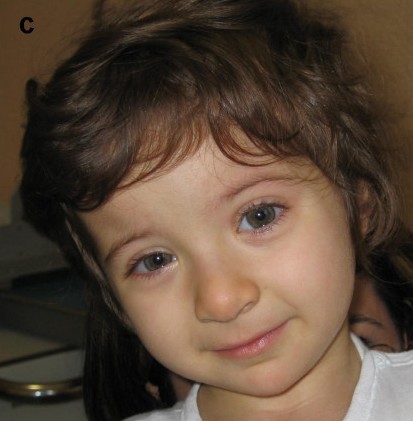
Rubinstein–Taybi syndrome (RTS) is a rare genetic condition characterized by short stature, moderate to severe learning difficulties, distinctive facial features, and broad thumbs and first toes.[2] Other features of the disorder vary among affected individuals. These characteristics are caused by a mutation or deletion in the CREBBP gene, located on chromosome 16, and/or the EP300 gene, located on chromosome 22.[3]
en.wikipedia.org
Rubinstein–Taybi syndrome (RTS) is a rare genetic condition characterized by short stature, moderate to severe learning difficulties, distinctive facial features, and broad thumbs and first toes.[2] Other features of the disorder vary among affected individuals. These characteristics are caused by a mutation or deletion in the CREBBP gene, located on chromosome 16, and/or the EP300 gene, located on chromosome 22.[3]
Typical features of the disorder include:
It is diagnosed when a heterozygous pathogenic variant of the CREBBP gene is identified in the individual. It exhibits an autosomal dominant inheritance pattern, but some documented cases show heterozygous individuals exhibiting germline mosaicism. This condition affects men and women equally, and is often misdiagnosed with other diseases or disabilities that result in delayed mental development.[citation needed]
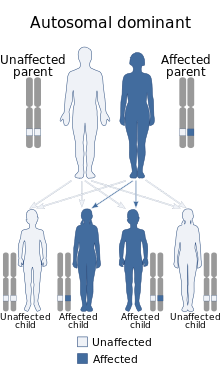 Rubinstein–Taybi syndrome is inherited in an autosomal dominant fashion.
Rubinstein–Taybi syndrome is inherited in an autosomal dominant fashion.
Rubinstein–Taybi syndrome, in many cases, is a microdeletion syndrome involving chromosomal segment 16p13.3 and is characterized by mutations in the CREBBP gene.[11][12] Varying amounts of material are deleted from this section of the chromosome and account for the spectrum of physiological symptoms.[13]
The CREBBP gene makes a protein that helps control the activity of many other genes. The protein, called CREB-binding protein, plays an important role in regulating cell growth and division and is essential for normal fetal development. If one copy of the CREBBP gene is deleted or mutated, cells make only half of the normal amount of CREB binding protein. A reduction in the amount of this protein disrupts normal development before and after birth, leading to the signs and symptoms of Rubinstein–Taybi syndrome.[14]
Mutations in the EP300 gene, located on chromosome 22q13.2, are responsible for a small percentage of cases of Rubinstein–Taybi syndrome.[3] These mutations result in the loss of one copy of the gene in each cell, which reduces the amount of p300 protein by half. Some mutations lead to the production of a very short, nonfunctional version of the p300 protein, while others prevent one copy of the gene from making any protein at all. Although researchers do not know how a reduction in the amount of p300 protein leads to the specific features of Rubinstein–Taybi syndrome, it is clear that the loss of one copy of the EP300 gene disrupts normal development.[citation needed]
CREBBP and p300 are the respective protein products of the paralogous genes CREBBP and EP300. Both of these related proteins, prototypical members of the p300-CBP coactivator family, have a bromodomain and a histone acetyltransferase domain and are able to bind to various gene-specific transcription factors as well as general transcription factors.[15] Cell lines derived from RTS patients exhibit diminished acetylation of multiple histone proteins, particularly histone 2A and histone 2B,[16] suggesting that this disease has its origins in problems with the regulatory mechanisms of transcription.[17] The functions of CREBBP and p300 broadly overlap but do have co-activator–specific effects on gene expression.[18] The proteins may also facilitate transcriptional elongation.[19]
A mouse model has been identified in order to perform experimental research procedures. The model has exhibited the same clinical symptoms seen in humans and has become a foundation for future research.[20]
Unfortunately, in approximately one third of cases, neither gene, CREBBP or EP300, can be implicated in the cause of the disease.[3]
Klinefelter syndrome - Wikipedia
The primary features are infertility and small, poorly functioning testicles (if present). These symptoms are often noticed only at puberty, although this is one of the most common chromosomal disorders, occurring in one to two per 1,000 live births.
The syndrome is defined by the presence of at least one extra X chromosome in addition to a Y chromosome yielding a total of 47 or more chromosomes rather than the usual 46.
Signs and symptoms[edit]


A person with typical untreated Klinefelter 46,XY/47,XXY mosaic, diagnosed at age 19 – a scar from biopsy is on his right breast above the nipple.
The Klinefelter syndrome has different manifestations and these will vary from one patient to another. Among the primary features are infertility and small, poorly functioning testicles. Often, symptoms may be subtle and many people do not realize they are affected. Whereas some other times symptoms are more prominent and may include weaker muscles, greater height, poor motor coordination, less body hair, gynecomastia (breast growth), and low libido. In the majority of the cases, these symptoms are noticed only at puberty.[10][6][4]
Prenatal[edit]
An estimated 60% of pregnancies with fetuses having Klinefelter syndrome spontaneously abort. Generally, the severity of the malformations is proportional to the number of extra X chromosomes present in the karyotype. For example, patients with 49 chromosomes (XXXXY) have a lower IQ and more severe physical manifestations than those with 48 chromosomes (XXXY).[14]Physical manifestations[edit]
As babies and children, those with XXY chromosomes may have weaker muscles and reduced strength. They may sit up, crawl, and walk later than other infants. In average KS children will start walking at 18 months of age. They also have less muscle control and coordination than other children of their age.[15]During puberty, they show less muscular body, less facial and body hair, and broader hips. This is a direct consequence of the low levels of testosterone produced by KS subjects. Delays in motor development may occur, which can be addressed through occupational and physical therapies. As teens, those assigned male at birth with XXY may develop breast tissue, have weaker bones, and a lower energy level than others. Testicles of those affected are usually less than 2cm in length (and always shorter than 3.5cm), 1cm in width, and 4ml in volume. Those with XXY chromosomes may also have microorchidism (i.e., small testicles).[15][16]
By adulthood, they tend to become taller than average; with proportionally longer arms and legs, less-muscular bodies, more belly fat, wider hips, narrower shoulders. Some will show little to no sign of affectedness, a lanky, youthful build and facial appearance, or a rounded body type with some degree of gynecomastia (increased breast tissue). Gynecomastia is present in approximately a third of affected individuals, a slightly higher percentage than in the XY population. Approximately 10% of those assigned male at birth with XXY chromosomes have gynecomastia noticeable enough that they may choose to have surgery. Those affected are often infertile, or have reduced fertility. Advanced reproductive assistance is sometimes possible in order to produce an offspring since approximately 50% of those assigned male at birth with Klinefelter syndrome can produce sperm.[17][18]
Psychological characteristics[edit]
Cognitive development[edit]
Some degree of language learning or reading impairment may be present, and neuropsychological testing often reveals deficits in executive functions, although these deficits can often be overcome through early intervention. It is estimated that 10% of those with Klinefelter syndrome are autistic. Additional abnormalities may include impaired attention, reduced organizational and planning abilities, deficiencies in judgment (often presented as a tendency to interpret non-threatening stimuli as threatening), and dysfunctional decision processing.[19][20]The overall IQ tends to be lower than average. Language milestones may also be delayed, particularly when compared to other people their age. Between 25% to 85% of those assigned male at birth with XXY have some kind of language problem, such as delay in learning to speak, trouble using language to express thoughts and needs, problems reading, and trouble processing what they hear. They may also have a harder time doing work that involves reading and writing, but most hold jobs and have successful careers.[15][21]
Behavior and personality traits[edit]
Compared to individuals with a normal number of chromosomes, those assigned male at birth affected by Klinefelter syndrome may display behavioral abnormalities. These are phenotypically displayed as higher level of anxiety and depression, mood dysregulation, impaired social skills, emotional immaturity during childhood or difficulty with frustration.[22][23][24] These neurocognitive abnormalities are most likely due to the presence of the extra X chromosome, as indicated by studies carried out on animal models carrying an extra X chromosome.[25]In 1995 a scientific study evaluated the psychosocial adaptation of 39 adolescents with sex chromosome abnormalities. It demonstrated that those assigned male at birth with XXY tend to be quiet, shy and undemanding; they are less self-confident, less active, and more helpful and obedient than other children their age. They may struggle in school and sports, meaning they may have more trouble fitting in with other kids.[21][26]
As adults, they live lives similar to others without the condition; they have friends, families, and normal social relationships. Nonetheless, some individuals may experience social and emotional problems due to problems in childhood. They show a lower sex drive and low self esteem, in most cases due to the feminine characteristics that their bodies display.[10][21]
Concomitant illness[edit]
Those with XXY are more likely than others to have certain health problems, such as autoimmune disorders, breast cancer, venous thromboembolic disease, and osteoporosis. Nonetheless the risk of breast cancer still below the normal risk for those assigned female at birth. These patients are also more prone to develop a cardiovascular disease due to the predominance of metabolic abnormalities such as dyslipidemia and type 2 diabetes. Interestingly, its has not been demonstrated that hypertension is related with KS.[27][28][29]In contrast to these potentially increased risks, rare X-linked recessive conditions are thought to occur less frequently in those with XXY than in those without, since these conditions are transmitted by genes on the X chromosome, and people with two X chromosomes are typically only carriers rather than affected by these X-linked recessive conditions.[30]
Epidemiology[edit]
This syndrome, evenly distributed in all ethnic groups, has a prevalence of approximately four subjects per every 10,000 (0.04%) males in the general population.[32][59][60][61] However, it is estimated that only 25% of the individuals with Klinefelter syndrome are diagnosed throughout their lives.[46] The rate of Klinefelter syndrome among infertile males is 3.1%. The syndrome is also the main cause of male hypogonadism.[62]
Rubinstein–Taybi syndrome - Wikipedia
Typical features of the disorder include:
- Broad thumbs and broad first toes and clinodactyly of the 5th finger[4]
- Mental disability
- Small height, low bone growth, small head
- Cryptorchidism in males
- Unusual facies involving the eyes, nose, and palate
- Anesthesia may be dangerous in these patients: "According to the medical literature, in some cases, individuals with Rubinstein–Taybi syndrome may have complications (e.g., respiratory distress and/or irregular heart beats [cardiac arrythmias]) associated with a certain muscle relaxant (succinylcholine) and certain anesthesia. Any situations requiring the administration of anesthesia or succinylcholine (e.g., surgical procedures) should be closely monitored by skilled professionals (Anesthesiologists)."[5] Primary literature suggests the children may have a higher rate of cardiac physical and conduction abnormalities which may cause unexpected results with cardioactive medications.[6] A further editorial reply in the British Journal of Anaesthesia discusses changes in the face and airway structure making it more difficult to secure the airway under anaesthesia, however, complications appeared in a minority of cases, and routine methods of airway control in the operating room appears to be successful. They recommended close individual evaluation of Rubinstein–Taybi patients for anaesthetic plans.[7]
It is diagnosed when a heterozygous pathogenic variant of the CREBBP gene is identified in the individual. It exhibits an autosomal dominant inheritance pattern, but some documented cases show heterozygous individuals exhibiting germline mosaicism. This condition affects men and women equally, and is often misdiagnosed with other diseases or disabilities that result in delayed mental development.[citation needed]
Genetics[edit]
 Rubinstein–Taybi syndrome is inherited in an autosomal dominant fashion.
Rubinstein–Taybi syndrome is inherited in an autosomal dominant fashion.Rubinstein–Taybi syndrome, in many cases, is a microdeletion syndrome involving chromosomal segment 16p13.3 and is characterized by mutations in the CREBBP gene.[11][12] Varying amounts of material are deleted from this section of the chromosome and account for the spectrum of physiological symptoms.[13]
The CREBBP gene makes a protein that helps control the activity of many other genes. The protein, called CREB-binding protein, plays an important role in regulating cell growth and division and is essential for normal fetal development. If one copy of the CREBBP gene is deleted or mutated, cells make only half of the normal amount of CREB binding protein. A reduction in the amount of this protein disrupts normal development before and after birth, leading to the signs and symptoms of Rubinstein–Taybi syndrome.[14]
Mutations in the EP300 gene, located on chromosome 22q13.2, are responsible for a small percentage of cases of Rubinstein–Taybi syndrome.[3] These mutations result in the loss of one copy of the gene in each cell, which reduces the amount of p300 protein by half. Some mutations lead to the production of a very short, nonfunctional version of the p300 protein, while others prevent one copy of the gene from making any protein at all. Although researchers do not know how a reduction in the amount of p300 protein leads to the specific features of Rubinstein–Taybi syndrome, it is clear that the loss of one copy of the EP300 gene disrupts normal development.[citation needed]
CREBBP and p300 are the respective protein products of the paralogous genes CREBBP and EP300. Both of these related proteins, prototypical members of the p300-CBP coactivator family, have a bromodomain and a histone acetyltransferase domain and are able to bind to various gene-specific transcription factors as well as general transcription factors.[15] Cell lines derived from RTS patients exhibit diminished acetylation of multiple histone proteins, particularly histone 2A and histone 2B,[16] suggesting that this disease has its origins in problems with the regulatory mechanisms of transcription.[17] The functions of CREBBP and p300 broadly overlap but do have co-activator–specific effects on gene expression.[18] The proteins may also facilitate transcriptional elongation.[19]
A mouse model has been identified in order to perform experimental research procedures. The model has exhibited the same clinical symptoms seen in humans and has become a foundation for future research.[20]
Unfortunately, in approximately one third of cases, neither gene, CREBBP or EP300, can be implicated in the cause of the disease.[3]





